RVD2: an ultra-sensitive variant detection model for low-depth heterogeneous next-generation sequencing data. Bioinformatics, 2015
Abstract
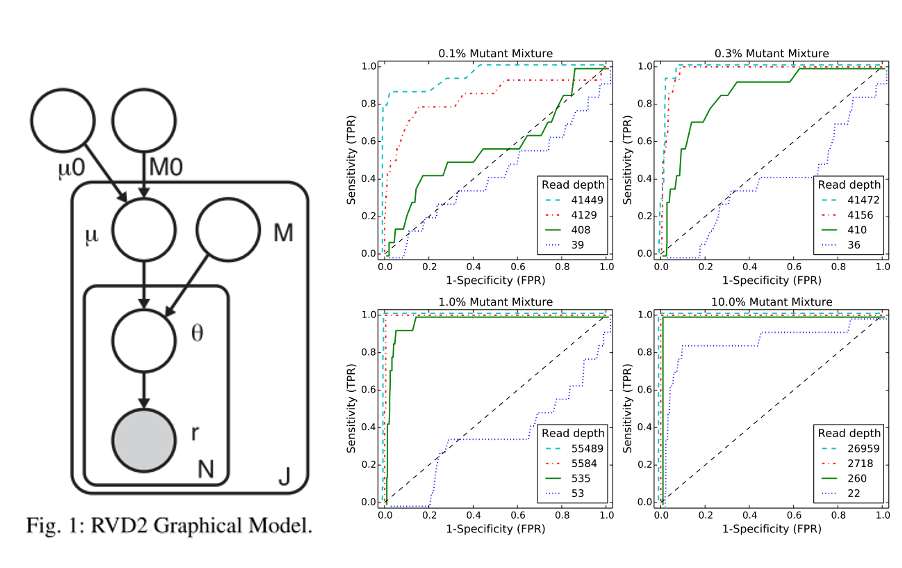
We present a novel variant calling algorithm that uses a hierarchical Bayesian model to estimate allele frequency and call variants in heterogeneous samples. We show that our algorithm improves upon current classifiers and has higher sensitivity and specificity over a wide range of median read depth and minor allele fraction. We apply our model and identify 15 mutated loci in the PAXP1 gene in a matched clinical breast ductal carcinoma tumor sample; two of which are likely loss-of-heterozygosity events.
Type
Publication
In Bioinformatics