Variational inference for rare variant detection in deep, heterogeneous next generation sequencing data. BMC Bioinformatics, 2017
Abstract
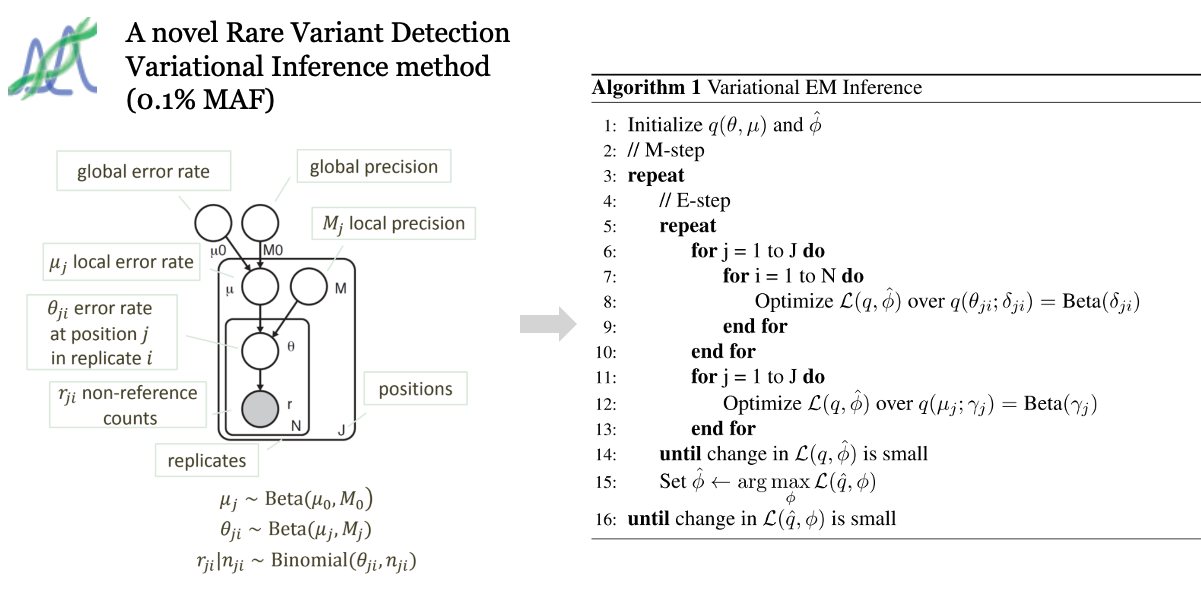
We propose a Bayesian statistical model and a variational expectation maximization (EM) algorithm to estimate non-reference allele frequency (NRAF) and identify SNVs in heterogeneous cell populations. We demonstrate that our variational EM algorithm has comparable sensitivity and specificity compared with a Markov Chain Monte Carlo (MCMC) sampling inference algorithm, and is more computationally efficient on tests of relatively low coverage (27× and 298×) data. Furthermore, we show that our model with a variational EM inference algorithm has higher specificity than many state-of-the-art algorithms. In an analysis of a directed evolution longitudinal yeast data set, we are able to identify a time-series trend in non-reference allele frequency and detect novel variants that have not yet been reported. Our model also detects the emergence of a beneficial variant earlier than was previously shown, and a pair of concomitant variants.